|
|
|
|
|
|
|
|
We provided crucial insights to the molecular mechanism through which HBV infects cells. A fusion peptide in preS1 and the human protein-disulfide isomerase ERp57 are involved in HBV membrane fusion process.
Link to the article
|
|
|
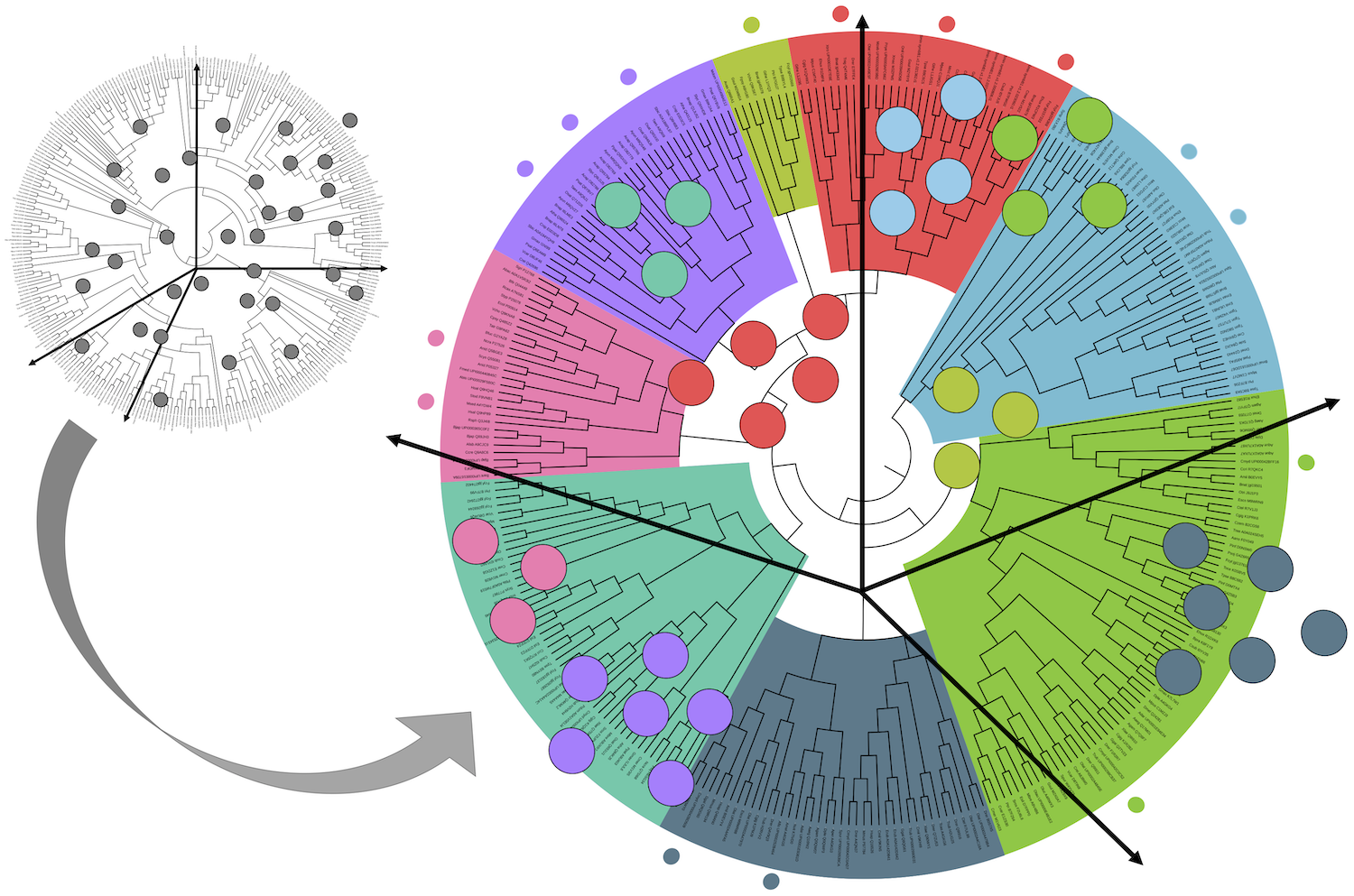
A global study of 60 cities' microbes finds each has a signature microbial fingerprint. The project provides a great way to communicate about the invisible world of commensal microorganisms. Members at the LCQB, L3 and M1 Sorbonne’s students involved in our Bioinformatics courses and colleagues from other Sorbonne departments contributed to the collect of the samples for the Paris area. The article just appeared in Cell.
|
|
|
|
|
|
|